定量蛋白质组学揭示艾滋病并患非霍奇金淋巴瘤的血浆蛋白变化
阅读:1838
时间:2023-01-06


前言
今天笔者分享的是发表在国际学术期刊Journal of Medical Virology(IF=20.693)上的一篇学术论文。该项目是由武汉大学病毒学国家重点实验室蓝柯教授团队和中南医院感染科桂希恩教授团队合作开展的研究,武汉大学庄柯博士和中南医院张永喜医师是该论文的共同第一作者,谱度众合(武汉)生命科技有限公司的黄邵鑫博士和陈希博士是该论文的共同作者。
本文运用蛋白质组学揭示了AIDS-NHL(Acquired immune deficiency syndrome-Non‐Hodgkin lymphoma) 相较于HIV (Human Immunodeficiency Virus)感染者的血浆分子变化,旨在为AIDS-NHL发病机制提供新的分子水平信息。
NHL是具有很强异质性的一组独立疾病的总称,是我国比较常见的肿瘤之一,在常见恶性肿瘤中位列前十,而HIV感染者发生淋巴瘤的风险大大增加,比普通人高约60 ~ 100倍,并倾向于发展出一种侵略性和治疗耐药表型,导致病情快速发展和预后不良。
实验设计:横断面研究
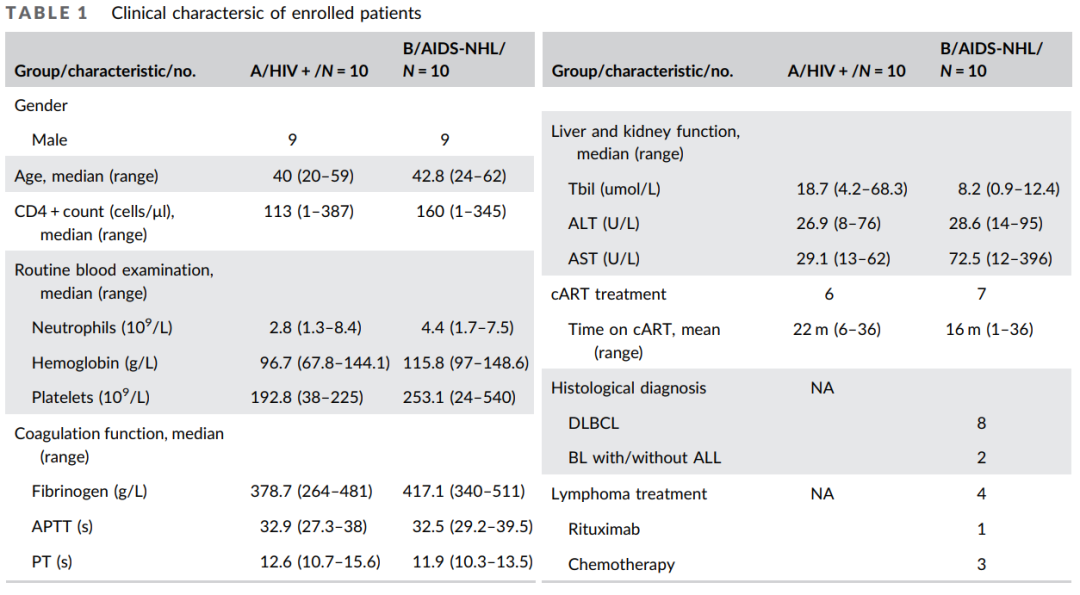
基线资料:20例HIV阳性患者的临床指标
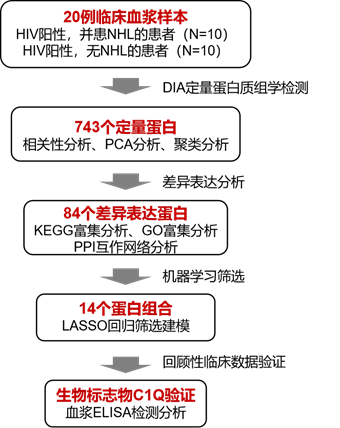
研究团队共采集了患有或不患有NHL的HIV感染者血液样本各10例。分离血浆后,通过DIA(Data‐independent‐acquisition)相对定量蛋白质组学进行分析,结合富集分析、PPI(Protein-protein network)以及LASSO回归筛选出目标蛋白,通过对患者病历中相关蛋白ELISA结果回顾统计,对目标蛋白进行验证。本文的发现揭示了AIDS-NHL发病机制的新的信息,并为AIDS-NHL相关的血浆生物标志物提供新的依据。
结果一:血浆蛋白质组学检测分析
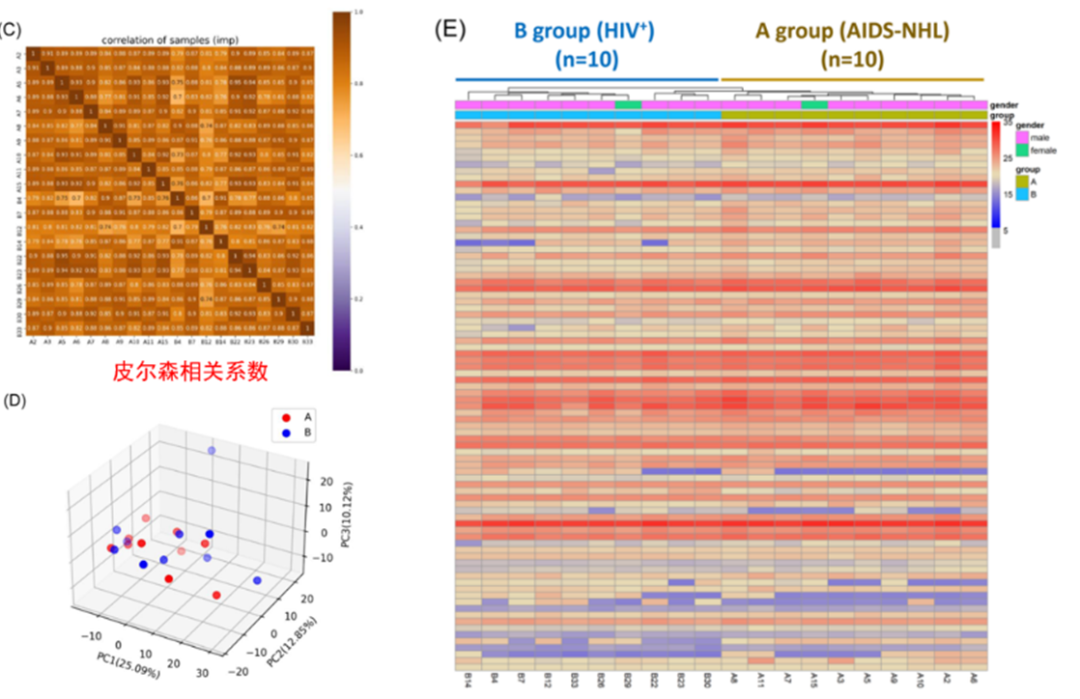
从收集的20例HIV和AIDS-NHL患者的样本中,通过DIA相对定量的扫描方式,总共鉴定到743个蛋白和6157个肽段。Pearson相关系数分析和主成分分析(Principal component analysis, PCA)表明样品重复性好,符合统计一致性。基于蛋白定量数据进行层次聚类分析和热图展示,表明部分蛋白在两组样品间具有表达差异,可能作为候选的蛋白分子标志物。
结果二:AIDS-NHL患者血浆蛋白质组特征
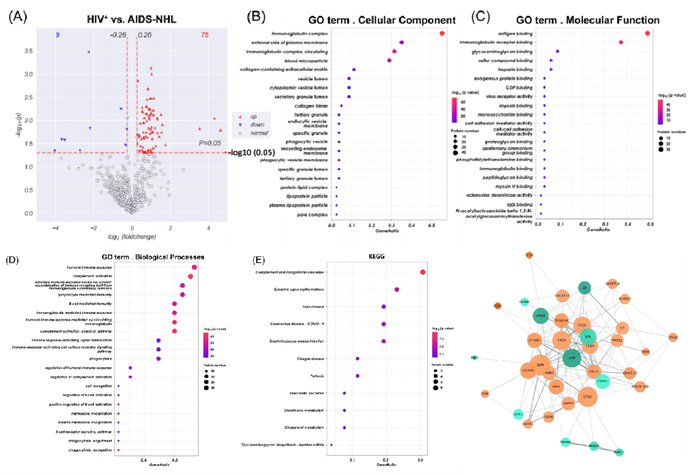
以|log2FC|>0.26,Adjusted P<0.05为标准,在AIDS和AIDS-NHL患者间筛选出84个表达量显著差异的蛋白(Differentially expressed proteins,DEPs)。通过对DEPs进行GO和KEGG注释和富集分析,发现这些蛋白几乎都参与了如体液免疫应答和补体激活等生物进程,其中极显著的3个补体蛋白富集于补体和凝血级联通路。PPI显示这些表达量变化显著的蛋白间普遍存在互作现象,其中β2-微球蛋白(β2‐microglobulin, B2M)在PPI网络中具有最高的相互作用程度,其次是组织蛋白酶D(Cathepsin, CTSD)和各种补体成分(C1Q子成分亚基)。
结果三:基于机器学习筛选蛋白分子标志物组合
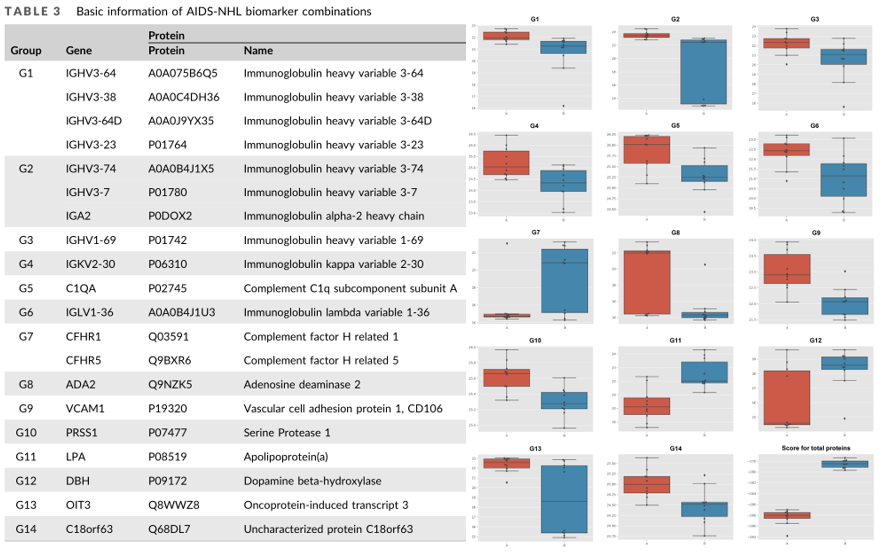
基于差异表达分析结果进一步将筛选获得的84个DEPs作为变量,使用R软件包“Glmnet”,通过最小绝对收缩和LASSO回归进行筛选和验证,建立DEPs的风险评分,最后构建优化模型。最终筛选得到14个蛋白集合,共20个蛋白成员组成的标志物候选蛋白集合。
结果四:C1Q作为蛋白分子标志物的ELISA验证
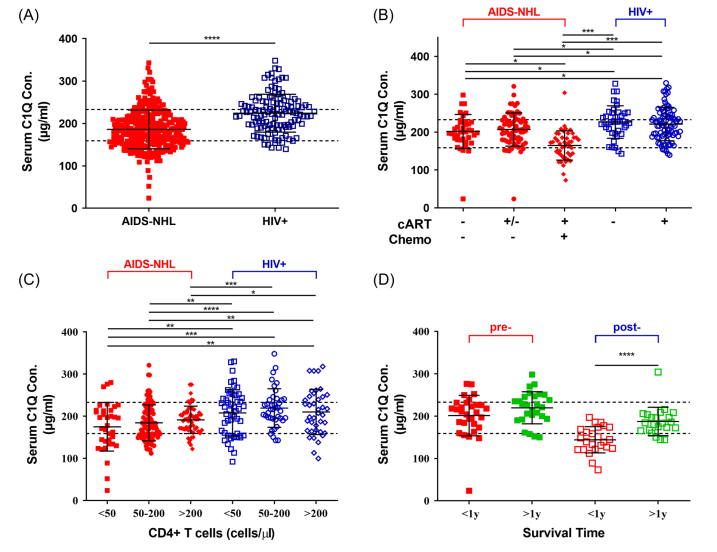
该研究对更广泛的患者诊断病历进行回顾性研究。共统计了79例AIDS-NHL患者和126例HIV感染者的血浆C1Q和β2M蛋白ELISA定量结果,发现AIDS-NHL患者血浆C1Q水平极显著低于对照组HIV感染者(图A),与蛋白质组学呈现一致性。此外,在AIDS-NHL患者中,生存时间<1年的AIDS-NHL患者C1Q水平均极显著低于生存时间>1年患者(P<0.001),在整个随访期间,大多数生存时间<1年的AIDS-NHL患者血浆C1Q水平下降趋势显著(图D)。结果表明,C1Q可能参与了AIDS‐NHL的病程发展,并有望作为一个潜在的生物标志物对AIDS‐NHL患者的预后进行预测。
结论
研究通过对20例患者进行血浆蛋白质组学分析,发现了20个具有潜在价值的AIDS-NHL蛋白生物标志物。其中C1Q在更大队列的病人病历的回顾性分析中得到了进一步的验证,可作为病程发展的潜在生物标志物进一步深入研究和验证。
总结
本文采取的横断面研究设计,能够为疾病的病因和预后提供线索,适合于一个研究方向的起始阶段。进一步的研究中,可增加发现队列的样本量,采用多种关键蛋白的建模筛选方式,并用更大独立队列进行验证,以提高证据力度。
从研究的临床意义上考虑,后续在较大样本下可将蛋白质的表达水平与临床数据结合,构建诊断模型,并对模型进行准确度、区分度和临床价值评估;或者在有条件的情况下,在HIV感染者病程早期取样并进行随访,观察是否并患NHL作为结局,构建疾病的预测模型,来寻找预测NHL疾病发生的蛋白质标志物,可能会有更大临床价值。

(研究总结表)


















