从姜黄素到奎宁经过14个案例拆解,我终于摸清了天然药物靶点筛选的适配逻辑!
阅读:1172
时间:2025-11-25
长期扎根于天然药物靶点筛选这块沃土,我总觉得技术选择就像 “给作物选农具”—— 没有绝对的 “最好”,只有 “适配场景的最优”。过去几年,从最初靠 DARTS 做低成本初筛,到用 CETSA 验证体内靶点,再到尝试 TPP 做全蛋白组探索,踩过不少坑,也积累了一些权威文献的实践经验。
今天就从实际研究出发,和大家聊聊 DARTS、SPROX、CETSA、TPP 这四种无标记技术在天然药物靶点筛选中的应用细节、各自的 “用武之地”,以及技术间如何搭配使用,希望能给各位研究者提供一些参考。
一、DARTS:天然药物靶点初筛的 “性价比之选”
第一次用 DARTS 是研究一种难修饰的黄酮类天然产物 —— 没有合适的活性基团做标记,亲和层析根本没法用,最后抱着试试的心态用了 DARTS,没想到真筛出了潜在靶点。现在回想,DARTS 能成为很多实验室的 “初筛标配”,核心在于它 “简单、便宜、不挑样品”。
1.核心逻辑与天然药物的适配性
● DARTS 的原理很直观:药物和靶点蛋白结合后,蛋白结构会更稳定,面对胰蛋白酶这类蛋白酶时,降解速率会变慢。我们只需要把天然药物(或粗提物)和细胞裂解液孵育,加蛋白酶消化后,通过 SDS-PAGE 检测 “没被降解的蛋白”,就能锁定潜在靶点。
这种 “不修饰药物” 的特性,对天然产物太友好了 —— 很多天然药物(比如萜类、生物碱)结构复杂,强行修饰会破坏活性,而 DARTS 直接用原生药物孵育,完全保留其生物活性。
【比如在研究姜黄素时,研究者用 DARTS 在 HCT116 细胞裂解液中孵育后,发现 STAT3 蛋白的降解量比对照组少 40%,后续通过 WB 和分子对接验证,确认 STAT3 是姜黄素抗结直肠癌的关键靶点,整个初筛过程成本不到 500 元。】
2.实际应用中的优势场景
● 粗提物初筛,快速缩小范围:研究中药复方或植物粗提物时,我们没法先分离单个成分,DARTS 可以直接用粗提物孵育样本,筛选出能被 “活性成分保护” 的蛋白。
【研究丹参粗提物时,用 DARTS 发现 PI3K 蛋白在提取物处理组的降解率显著降低,后续分离出丹酚酸 B 验证,确认丹酚酸 B 是结合 PI3K 的主要活性成分,比先分离再筛选节省了 2 个月时间。】
● 捕捉低亲和力靶点,避免漏检:天然药物常存在 “多靶点弱结合” 的特点,传统亲和层析需要强亲和力才能 “拉” 住靶点,容易漏检低亲和力靶点。而 DARTS 没有洗涤步骤,即使药物与靶点是弱相互作用,也能通过 “抗降解” 信号检测到。
【比如白藜芦醇通过 DARTS 筛选时,除了已知的高亲和力靶点 SIRT1,还捕捉到了低亲和力靶点 eIF4A(降解率降低 25%),后续功能实验证明 eIF4A 在白藜芦醇抑制肿瘤转移中起重要作用。】
3.不得不注意的局限
● DARTS 的短板也很明显:低丰度蛋白难检测 —— 如果靶点蛋白在细胞中含量低,即使被药物保护不降解,凝胶上也难显影。
【我之前研究一种生物碱时,DARTS 没检测到低丰度的膜蛋白,后续用 CETSA 才确认其结合;另外,膜蛋白本身对蛋白酶消化的抵抗力较强,可能会被误认为是 “药物保护的靶点”,需要后续用其他技术交叉验证。】
二、CETSA:天然药物靶点 “体内验证的金标准”
如果说 DARTS 是 “初筛探路者”,那 CETSA 就是 “验证把关人”。我第一次用 CETSA 是为了确认 DARTS 筛出的靶点是否在体内真的结合 —— 体外实验做得再好,体内不结合也没用,而 CETSA 恰好能解决 “体内外差异” 这个痛点。
● CETSA 的核心是 “热稳定性变化”:未结合药物的蛋白加热到一定温度(比如 50℃)会变性沉淀,结合药物的蛋白热稳定性提升,同样温度下仍能溶解。通过 Western blot或免疫荧光检测溶解液中的蛋白,就能判断药物是否与靶点在细胞 / 组织中结合。
这种 “在生理环境中验证” 的特性,对天然药物研究至关重要。
【研究抗肝癌生物碱 HTBPI 时,先用化学 proteomics 推测靶点是 AKT,但体外结合实验不能证明体内也结合;后来用小鼠肝癌组织做 CETSA—— 加热到 45℃时,HTBPI 处理组的 AKT 溶解量是对照组的 2.3 倍,直接证明了 HTBPI 在体内能结合 AKT,这是体外技术无法替代的。】
2.实际应用中的关键价值
● 体内靶点验证,贴近临床场景:天然药物最终要用于体内,体外筛选的靶点常与体内情况脱节。CETSA 能直接用完整细胞、动物组织甚至患者活检样本做实验,结果更贴近实际疗效。
【研究青蒿素抗疟时,用恶性疟原虫感染的红细胞(完整细胞)做 CETSA,发现青蒿素处理组的 MD2 蛋白在 57℃仍能溶解,对照组则沉淀,证明青蒿素在疟原虫感染的红细胞中确实结合 MD2,为后续临床剂量设计提供了依据。】
● 量化药物效能,指导剂量优化:CETSA 的 “等温剂量反应(ITDR-CETSA)” 模式能计算 EC50 值,评估药物结合靶点的 “ 效能 ”。
【研究藤黄酸(抗胰腺癌天然产物)时,ITDR-CETSA 显示其结合 Bcl-2 的 EC50 是 20μM,据此设计动物实验剂量(15mg/kg),既保证了靶点结合率,又避免了毒性,比盲目试剂量节省了大量动物资源。】
3.实际操作中的挑战
● 低通量:一次 WB-CETSA 只能检测 1-2 个靶点,要是想验证 TPP 筛出的十几个潜在靶点,得跑十几轮实验。
● 依赖高质量特异性抗体:如果靶点蛋白没有商业化抗体(比如一些新发现的转录因子),就只能放弃,这在天然药物筛选中很常见。
三、SPROX:天然药物 “结合机制解析的利器”
1.核心逻辑与天然药物机制研究的适配性
● SPROX 基于“甲硫氨酸氧化速率变化”:蛋白中的甲硫氨酸残基容易被氧化,药物与靶点结合后会改变蛋白空间结构,进而影响甲硫氨酸的氧化速率 —— 结合药物的蛋白,甲硫氨酸氧化变慢(或变快)。通过量化氧化比例,不仅能判断是否结合,还能算出结合热力学参数(如 ΔG),解析结合机制。这种 “量化结合特性” 的能力,对天然药物机制研究很重要。
【研究环孢素 A(天然免疫抑制剂)时,通过SPROX 不仅确认了已知靶点亲环蛋白 A,还算出结合自由能 ΔG,同时发现新靶点 GalE,为解释环孢素 A “多靶点协同免疫抑制” 提供了热力学依据。】
2.实际应用中的独特价值
● 补充验证,排除间接作用:有时候 DARTS 发现蛋白 “抗降解”、CETSA 发现 “耐热”,但不确定是药物直接结合还是间接作用(比如下游信号激活导致蛋白稳定)。SPROX 能通过检测甲硫氨酸氧化变化,确认是直接结合导致的稳定性变化。
【研究小檗碱时,DARTS 显示其保护 AMPK 不降解,SPROX 进一步发现 AMPK 的甲硫氨酸氧化率降低 30%,直接证明小檗碱与 AMPK 的直接结合,排除了 “下游信号激活” 的干扰。】
● 适配定量蛋白质组学,提升通量:传统 SPROX 通量低,但升级后的 SILAC-SPROX(稳定同位素标记结合 SPROX)能同时分析多个蛋白。
【用 SILAC-SPROX 研究茶多酚提取物,一次检测了 200 多个含甲硫氨酸的蛋白,发现 EGCG 能特异性降低 ALDH1A3 的甲硫氨酸氧化速率,后续验证其是 EGCG 抗肝癌的关键靶点,通量比传统 SPROX 提升了 10 倍。】
3.应用中的限制因素
● 依赖甲硫氨酸:如果靶点蛋白不含甲硫氨酸(虽然少见,但确实存在),SPROX 就无法检测。
● 药物用量大:需要 mM 级浓度,而很多天然产物溶解度低,比如某些萜类化合物,很难达到这个浓度,限制了其应用场景。
四、TPP:天然药物靶点 “全域探索的新选择”
聊完前三种技术,再说说 TPP。我第一次用 TPP 是为了探索一种中药复方的 “多靶点谱”—— 前三种技术要么低通量,要么需要预设靶点,没法满足 “全蛋白组无偏向筛选” 的需求,而 TPP 恰好填补了这个空白。
1.核心逻辑与天然药物多靶点研究的契合
● TPP 可以理解为 “CETSA 的高通量升级版”:同样基于蛋白热稳定性变化,但结合了质谱技术(如 TMT 标记),一次实验能检测细胞或组织中上千种蛋白的热稳定性变化,无需预设靶点,实现全蛋白质组层面的靶点筛选。这种 “无偏向、高通量” 的特性,完美适配天然药物 “多靶点、复杂机制” 的特点。
【研究放线菌来源的天然产物十字孢碱时,TPP 在 K562 细胞裂解液中一次检测了 7000 + 蛋白,不仅筛选出 51 个已知的激酶靶点,还发现了 2 个意外靶点 —— 粪卟啉原 III 氧化酶和亚铁螯合酶,这是之前用 DARTS、CETSA 无法实现的,为其药理机制研究开辟了新方向。】
2.与前三种技术的协同与突破
TPP 不是 “取代” 前三种技术,而是 “互补升级”:
● 突破 “预设靶点” 局限:DARTS、CETSA、SPROX 都需要先有 “潜在靶点列表”,而 TPP 无需预设,能发现未知靶点。
【研究海洋天然产物 13²- 羟基脱镁叶绿素 a 时,ABPP 技术(需修饰)没找到靶点,TPP 却在 HepG2 细胞中发现 9 个脂质调节相关靶点,包括 RPSA、FGG 等新靶点,后续功能实验证明这些靶点协同调控脂质代谢。】
● 解决 “高通量” 痛点:DARTS、CETSA、SPROX一次最多分析几十个蛋白,而 TPP 一次能分析上千个。
【研究中药白术的活性成分 atractylenolide I 时,TPP 一次就发现了 PSMD4、CGGBP1、SORBS3 三个靶点,后续用 CETSA 逐一验证,效率比单独用 CETSA 提升了 3 倍。】
● 适配复杂样本,解析全景机制:TPP 能在组织样本中做全蛋白组筛选,不仅能找到直接靶点,还能发现下游蛋白变化。
【研究抗疟药奎宁时,TPP 在恶性疟原虫感染的红细胞中,不仅找到直接靶点 PfPNP,还发现了下游嘌呤代谢相关蛋白(如 HGPRT)的热稳定性变化,完整解析了奎宁 “抑制嘌呤合成” 的抗疟机制。】
3.实际应用中的注意事项
数据分析需要专业工具(如 R 包 TPP、ProSAP 软件),需要一定的生物信息学基础。但对于 “发现新靶点、解析多成分复方” 这类核心需求,这些投入是值得的。
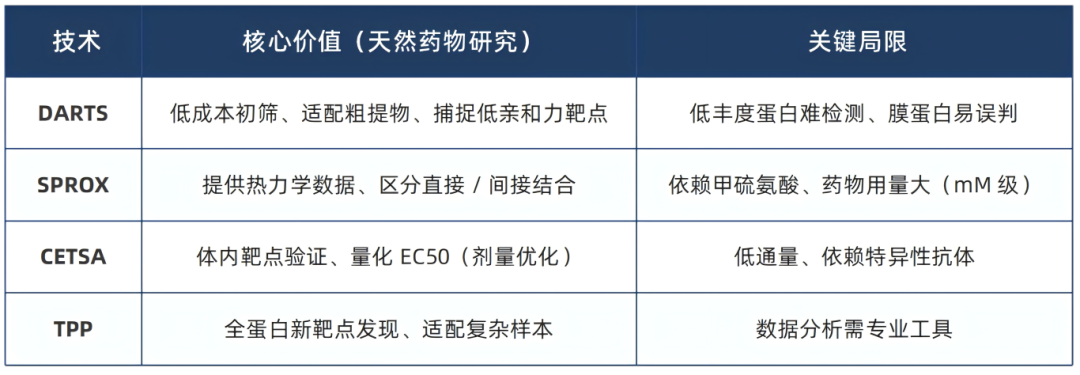
五、技术核心对比
六、结语:技术选择的核心是 “适配研究目标”
这些无标记技术的发展为天然药物靶点筛选提供了“不破坏天然产物活性、贴近生理场景、适配多靶点特性” 的解决方案。其中,DARTS 与 SPROX 在初筛与机制解析中不可或缺,CETSA 是体内验证的关键,而 TPP 则凭借高通量、无预设靶点的优势,成为天然药物 “新靶点发现与复杂机制解析”的核心推动力。
作为研究者,我们需明确:技术选择应紧扣研究目标 —— 初筛选 DARTS,机制解析选 SPROX,体内验证选 CETSA,新靶点探索选 TPP。唯有基于文献指导的技术搭配,才能高效、准确地挖掘天然药物的靶点价值,为后续药物开发奠定基础。
推荐
谱度众合:最经典、主流的药物靶点筛选技术——TPP热蛋白组分析
✔ 全面直接筛选真实药靶组合:蛋白质组水平筛选药物结合的蛋白靶点,全面覆盖治疗靶点与脱靶靶点;使用药物分子本体进行试验,无需设计合成分子探针,药靶结合更真实
✔ 多种数据分析策略:结合蛋白热变性曲线分析和非参数分析方法(NPARC),全面捕获潜在药物靶点
✔ 多种生信分析数据库挖掘辅助筛选:对潜在药物靶点进行生信分析与数据库挖掘,辅助最终药物靶点的确认
✔ 多种衍生技术可选:除常规温度范围(TPP-TR)、药物浓度范围(TPP-CCR)、两者结合(2D-TPP)的常规热蛋白组分析方法外,还可进行单温度点(ITSA)、多温度点混合(PISA)等高通量热蛋白组分析方法

















